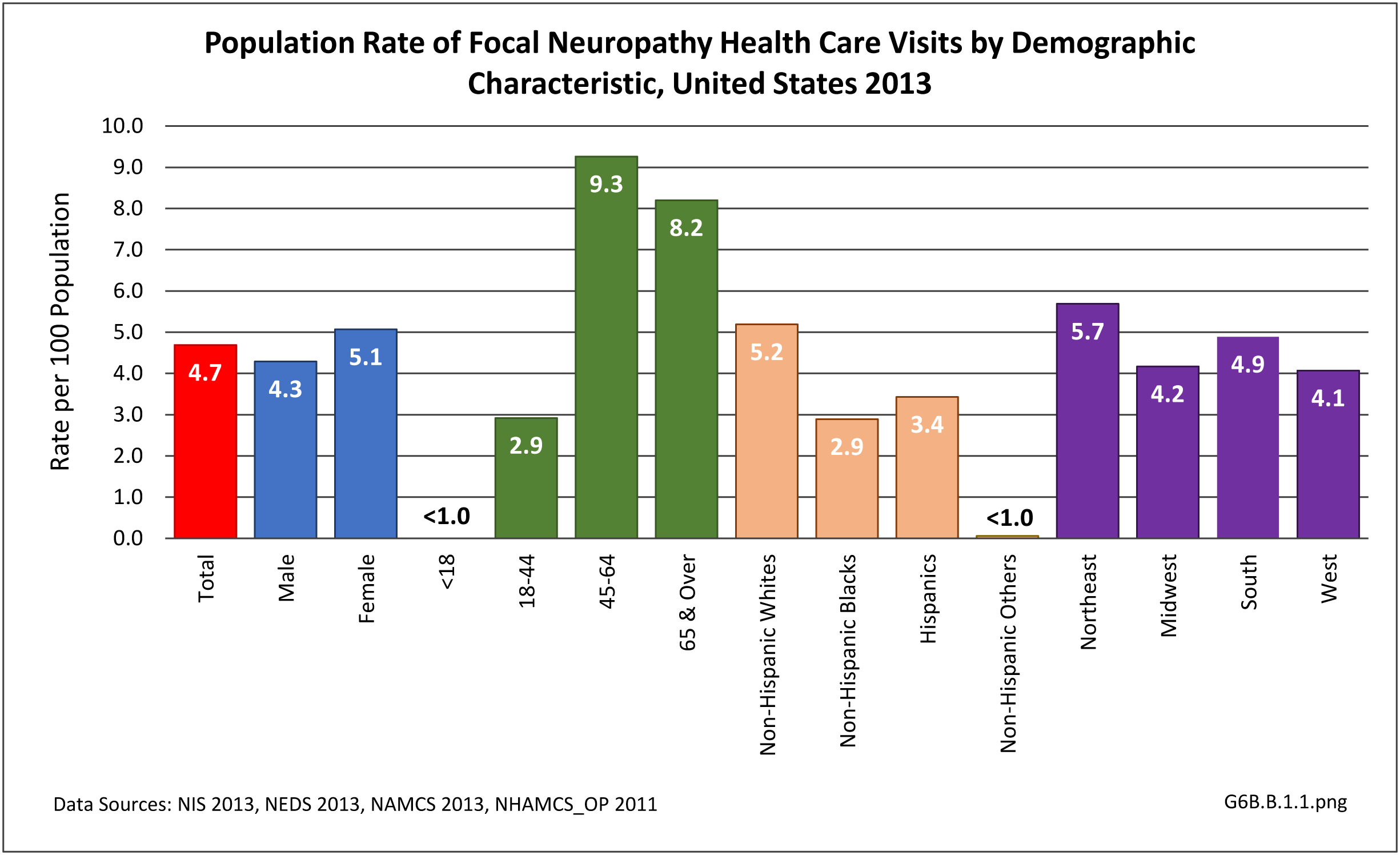
 [11]
[11] Neuromuscular disorder (NMD) is a collective term used to describe diseases that affect any part of the nervous system and muscles. Although there are many different forms that vary in onset, severity, and prognosis, NMDs can have a significant direct and indirect impact on an individual leading to loss of functional capacity.1
Different classification systems are available for neuromuscular disorders based on the location of involvement, the etiology, or presenting symptom. Based on the anatomic location of involvement, NMD can be categorized into
Based on the presenting symptoms, NMDs can be classified into disorders with sensory impairment, motor impairment, or both. Neuromuscular disorders can also be categorized roughly into hereditary or acquired neuromuscular NMDs.2 The breakdown of classifications based on the type of impairment at presentation include the following categories.
Sensory impairment can be divided into negative symptoms and positive symptoms. Negative symptoms, which include numbness and loss of joint proprioception with unsteadiness, are often prominent in hereditary conditions such as Charcot Marie Tooth disease or severe neuropathy. Generally, the positive sensory symptoms, such as tingling, pins/needle sensation, and burning pain, are more commonly recognized by patients and the reason they seek medical attention.
Peripheral neuropathy is the most common cause of sensory impairment. It can be divided into hereditary (often related to family history and genetic mutation) and acquired neuropathy. Depending on the area of involvement, neuropathy also can be classified into polyneuropathy, meaning multiple nerves are involved (e.g. distal symmetric polyneuropathy, brachial plexopathy), or mononeuropathy, such as carpal tunnel syndrome, cubital tunnel syndrome, or single level radiculopathies from a disc herniation or spinal stenosis.
The most common acquired polyneuropathy is secondary to diabetes, which affects an estimated 50% of the older type 2 diabetic population.3,4 The most common mononeuropathy is carpal tunnel syndrome (median nerve entrapment at the wrist), followed by ulnar nerve entrapment at the elbow. Charcot Marie Tooth disease is the most common hereditary neuropathy and has variable sensory involvement.
Motor impairment, manifested as muscle weakness and fatigue, can be secondary to motor neuron disease (such as amyotrophic lateral sclerosis), peripheral neuropathy (involving motor nerves), neuromuscular junction disorders (like myasthenia gravis), and myopathy. When it occurs with sensory impairment, peripheral neuropathy is the most common underlying cause.
Muscle pain (myalgia) or cramps are rare manifestations of some myopathies. Each group of disorders can be further classified depending on the location of involvement and underlying etiology.
Muscle weakness can be due to pain inhibition (from musculoskeletal conditions like arthritis). In contrast to the weakness accompanying pain, the weakness seen with neuromuscular disorders is more profound and often more progressive.
Swallowing difficulty (dysphagia), speech problems (dysarthria), and drooping of upper eyelid (ptosis) can be manifestations of muscle weakness in the head and neck secondary to motor neuron disease, myopathy, or neuromuscular disorders.
Mobility involves various types of movement, such as transferring from sitting to standing, walking, and stair/ramp negotiation. Motor impairments involving thigh and hip muscles limit the ability to stand, and can be caused by myopathy or neuropathy (lumbar plexopathy, amyotrophy, radiculopathy). Steady gait requires good balance and joint proprioception (the sense of the relative position of one's own parts of the body and strength of effort being employed in movement), which can be compromised in peripheral neuropathy with sensory impairment. Impairments of sensory and motor nerve functions are important risk factors for falls in older persons and relate directly to disability.5 If the distal muscles that cross the ankle are involved because of peripheral neuropathy or lumbosacral radiculopathy, patients may present with difficulty clearing their foot while walking, resulting in a dragging or slapping gait or “drop foot”.
Upper extremity muscle strength and sensation are important for activities of daily living (ADL), therefore, impairments can cause significant disability. Difficulty with dressing, brushing teeth and combing hair can occur with weakness of the shoulder and arm caused by myopathy, brachial plexopathy or radiculopathy. Injury to the hand and wrist from carpal tunnel syndrome, radiculopathy, motor neuron disease, etc. can lead to dropping objects, difficulty with buttoning and other fine motor skills.
A focal neuropathy means only one or, at most, a few nerves are injured. Pain, numbness, and weakness are confined to a single limb or a small region of the trunk or head. Focal neuropathies are typically caused by compression or trauma. Carpal tunnel syndrome is an example of a focal neuropathy (as described below).
Mononeuropathy is a form of neuropathy that affects a single nerve or, more rarely, a nerve group (mononeuritis multiplex). There may or may not be pain, followed by loss of sensation, strength, and overall function, depending on the type. These types of neuropathies are typically due to injury, compression, aging, inflammatory disorders, or other systemic diseases. Examples of mononeuropathy include carpal tunnel syndrome, ulnar neuropathy, trigeminal neuralgia, radial neuropathy, peroneal neuropathy, radiculopathy, and occipital neuralgia.
Mononeuropathy can develop if there has been a prolonged period of swelling or pressure placed on a specific point in the body such as the hands, feet, or face. Symptoms of mononeuropathy include loss of feeling, tingling, burning, muscle weakness, and paralysis.
Mononeuropathy can develop if there has been a prolonged period of swelling or pressure placed on a specific point in the body such as the hands, feet or face. Symptoms of mononeuropathy include loss of feeling, tingling, burning, muscle weakness, and paralysis.
Carpal tunnel syndrome
Carpal tunnel syndrome is the most common mononeuropathy and is caused by entrapment of the median nerve in the carpal tunnel at the wrist. It is a slowly progressive condition causing tingling, numbness, and pain in the hand and fingers (possibly sparing the pinky finger), with weakness and wasting of muscle at the base of the thumb. Based on different studies, the incidence varies between 0.99 to 3.5 persons per 1,000 person-years.1,2 Higher incidence has been reported in women and working populations requiring repetitive wrist motions. Carpal tunnel syndrome is diagnosed based on clinical information (history and physical examination) and confirmed by electrodiagnostic tests (EDx) consisting of nerve conduction studies (NCS) and electromyography (EMG).
Ulnar neuropathy
Ulnar neuropathy is the second most common entrapment neuropathy and most commonly occurs at the elbow. It presents with weakness of the hand, along with tingling, numbness, and pain in the inner side of the hand and fingers (half of ring finger and pinky). It is often triggered from irritation of the “funny bone” where the nerve is exposed along the inner aspect of the elbow.
The brachial plexus is a network of nerves that originate in the neck and branch off to form most of the other nerves that control movement and sensation in the upper limbs, including the shoulder, arm, forearm, and hand. The radial, median, and ulnar nerves originate from the brachial plexus.
Brachial plexus injury (BPI) is an umbrella term for a variety of conditions that may impair function of the brachial plexus nerve network. Most pediatric and adult brachial plexus injuries are caused by birth or trauma respectively, such as high-speed vehicular or motorcycle accidents, blunt trauma, stab or gunshot wounds. It can also be the result of inflammatory processes, compression (e.g., caused by a growing tumor, thickened muscles, or the collar bone), or genetic mutation. Symptoms are pain, loss of sensation, muscle weakness, and varying degrees of paralysis.
Radiculopathy is a nerve root disorder that can cause numbness, tingling, pain, and weakness. It is typically caused by acute or chronic pressure on a nerve root as it exits the spinal canal. The most common cause is a herniated intervertebral disc in younger patients, and spinal stenosis, or narrowing of the spinal canal, in older patients. There are also several less common causes such as meningitis, tumors, diabetes, and infections.
Cervical radiculopathy typically causes pain radiating form the neck to the arm or shoulder blade region., often accompanied by tingling, numbness or weakness. The prevalence of cervical radiculopathy is 3.3 cases per 1000, with average age-adjusted incidence rate of .8 cases per 1000 persons. Lumbosacral radiculopathy typically presents as “sciatica”, pain radiating from the lower back into the buttock, thigh and/or leg. It is more common than cervical radiculopathy with a prevalence of 3% to 5% of the adult population, which is evenly distributed between men and women.3 Most radiculopathies related to disc herniation are self-limiting with symptoms resolving over the course of weeks to months.4
Carpal tunnel syndrome is the most common reason for electrodiagnostic test (EDx) referral.1
Conservative management includes activity modification, wrist braces, occupational therapy, and ultrasound guided steroid injections, . Carpal tunnel release to decompress the entrapped nerve is the primary surgical treatment and is commonly done if symptoms persist despite conservative management. The cost of treatment varies widely depending on the type of surgery, surgery setting, and amount of occupational therapy or lost work. Surgery is typically performed in an outpatient setting, and can be done open, with a camera or with a small incision under ultrasound. Potential complications include scarring, recurrent symptoms or nerve damage, although surgery is typically successful after appropriate diagnostic workup and conservative treatment.
Patients with radiating neck and back pain and neurologic deficits often require diagnostic studies such as MRIs and EDx to confirm the clinical impression, to investigate the underlying cause, and to determine severity. Although most radiculopathies respond to conservative management (oral pain medication, physical therapy, image guided epidural steroid injections), some patients require surgical treatment to decompress the nerve root.
During 2013, there were more than 14.8 million healthcare visits made that included a diagnosis of focal neuropathy, representing nearly 1 in 20 persons in the US. Nearly all were adults over the age of 18, with most age 45 or over. Females were more likely to have a healthcare visit for focal neuropathy than males (55% of visits versus 45%), as were non-Hispanic whites. Non-Hispanic others accounted for less than 1% of focal neuropathy healthcare visits. Visits by geographic region were representative of the population. (Reference Table T6B.1.1 PDF [2] CSV [3]; Table 6B.1.2 PDF [4] CSV; [5] Table 6B.1.3 PDF [6] CSV [7]; and Table 6B.1.4 PDF [8] CSV [9])
[11] Slightly more than 1.2 in 100 health care visits in 2013 had a focal neuropathy diagnosis. Most of these visits (84%) were to a physician’s office, where the rate of focal neuropathy diagnoses was nearly 1.4% of all visits. Only 3% of the total health care visits with a focal neuropathy diagnosis were hospital discharges, yet this accounted for 427,100 discharges. A diagnosis of root neuropathy was made in 3 out of 4 diagnoses. (Reference Table 6B.1.5 PDF [12] CSV [13]) (G6B.B.1.2)
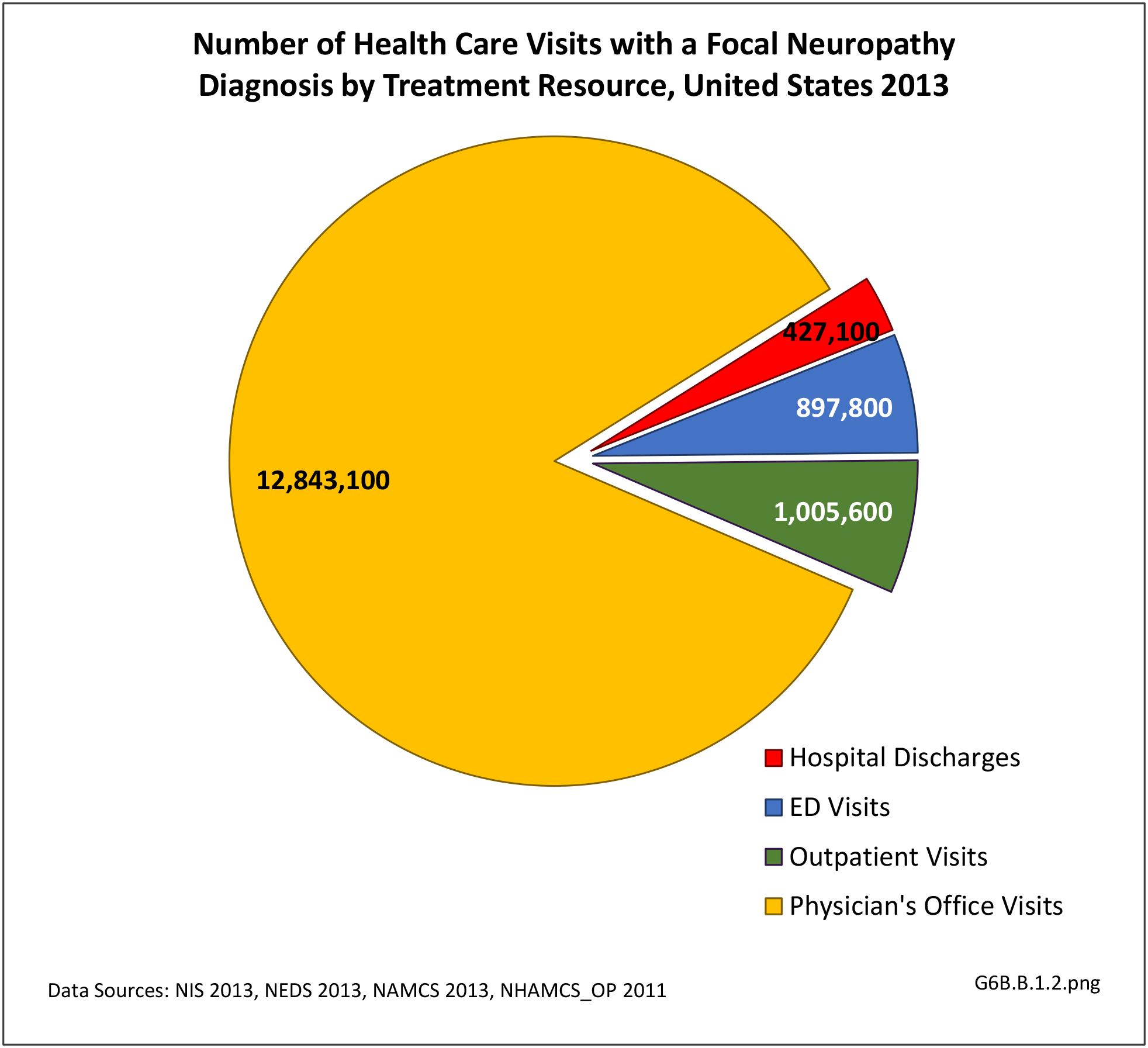 [15]
[15]
As most entrapment neuropathies and radiculopathies increase with age, the burden of these conditions is increasing with the aging demographic profile of the US. The elderly were reported to have a higher prevalence of severe carpal tunnel syndrome.1 Due to age-related changes in the spine, the underlying etiologies of radiculopathy are different among various age groups. For example, radiculopathy from disc herniation is common in young adults, whereas spinal stenosis and spondylosis are more common in the elderly population.2
The burden of entrapment neuropathies and radiculopathy includes direct health care costs related to diagnosis, physical and occupational therapy, pain management, surgical intervention, plus indirect costs due to lost works days and productivity. Carpal tunnel syndrome is one of the most common worker’s compensation diagnoses. Radiculopathy from neck or low back conditions also significantly hampers mobility, activities of daily living, and quality of life.
Mean hospital charges for an average stay of 4.3 days in 2013 were $64,400, although this would not be actual cost paid due to variations in actual payments. Also, the hospital charges did not include professional fees and non-covered charges, such as lab tests. Adding to the cost for about one-third (36%) of discharges with a diagnosis of focal neuropathy discharged from a hospital in 2013 was “discharged to additional care”, either at a skilled nursing facility or with home health care. Mean emergency department charges were $3,200 with a focal neuropathy diagnosis, with 1 in 5 (19%) cases admitted to the hospital. (Reference Table 6B.2.1 PDF [16] CSV [17] and Table 6B.2.2 PDF [18] CSV [19])
Peripheral neuropathy is a condition that develops from a dysfunction of the nerves that transmit motor or sensory information to and from the brain, spinal cord, and the rest of the body. An estimated 20 million people in the United States have some form of the more than 100 types of peripheral neuropathy.1,2 Peripheral neuropathy can be categorized as hereditary or acquired, with diabetes mellitus the most common cause of acquired peripheral neuropathy.1 Alcohol abuse is also a common cause.
Up to 70% of patients with diabetes eventually develop peripheral neuropathy, with symptoms ranging from subtle or no symptoms to tingling, pain, and profound weakness. Symptoms usually involve the longer nerves, affecting the toes, feet, then gradually progress up the body. Sensory symptoms are more common than motor, and pain occurs in 40-60% of patients with documented neuropathy.3
Guillan-Barre syndrome (GBS) is a peripheral neuropathy in which the body’s immune system attacks part of the peripheral nerve. The syndrome is not uncommon, afflicting approximately one person in 100,000. Usually GBS occurs a few days or weeks after a respiratory or gastrointestinal viral infection. Less commonly it occurs following surgery, or vaccination. There has recently been an increased incidence of GBS due to infection from the Zika virus.
Charcot-Marie-Tooth (CMT) is one of the most common inherited peripheral neuropathies, affecting 1 in 2500 people in the United States. It has several forms affecting different parts of the peripheral nervous system. Despite its inherited fashion, the onset of symptoms varies depending on the type and severity, and it can present anytime from childhood to adulthood. Symptoms may involve the foot, lower leg and hand/finger with numbness, tingling, weakness and muscle wasting. Due to weakness, fatigue and impaired gait, quality of life is significantly impaired.4 Some patients with severe involvement are disabled.5
The diagnosis of peripheral neuropathy is made based on clinical presentation, EDx, blood tests, and occasionally biopsy. Genetic testing is utilized in hereditary conditions, especially for women in their reproductive years, and for affected family members.1
There is no cure for most peripheral neuropathies, and health care resources are typically utilized for symptomatic treatment in outpatient settings, e.g., pain management, rehabilitation (including physical and occupational therapy), and bracing. Patients with diabetic neuropathy utilize greater healthcare resources and have higher costs than patients with diabetes without neuropathy.2
Nearly 6.4 million health care visits in 2013 had a diagnosis of peripheral polyneuropathy, representing 2 in 100 persons in the US. Age is a factor in polyneuropathy, with half (52%) the diagnoses occurring in the 65 and over population. Although males and females had similar rates of diagnosis, females were slightly more likely to have a polyneuropathy diagnosis than males. Because of the small number of diagnoses overall, it is difficult to determine racial/ethnic differences. The western region of the US had a higher share of diagnoses for peripheral polyneuropathy than expected based on population.(Reference Table T6B.1.1 PDF [2] CSV [3]; Table 6B.1.2 PDF [4] CSV [5]; Table 6B.1.3 PDF [6] CSV [7]; and Table 6B.1.4 PDF [8] CSV [9])
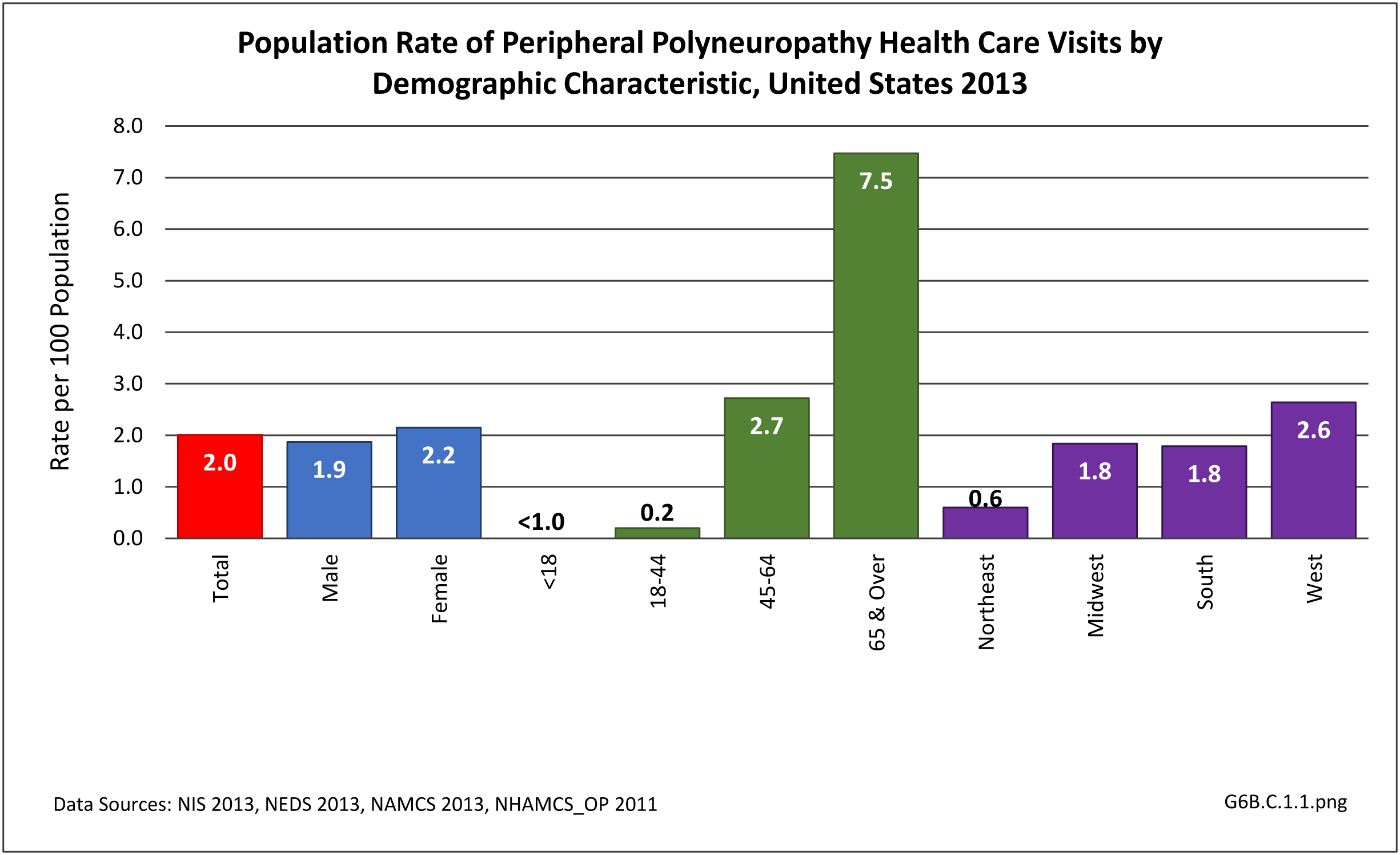 [21]
[21] Using the definition of hereditary polyneuropathy versus acquired, diagnoses were evenly split between the two types. While visits to a physician’s office represented more than half (58%) of all health care visits with a peripheral polyneuropathy diagnosis in 2013, the share of hospital discharges with this diagnosis was much higher than that of physician office visits (3.0/100 versus 0.4/100). More than one million hospital discharges had a polyneuropathy diagnosis in 2013. (Reference Table 6B.1.5 PDF [12] CSV [13])
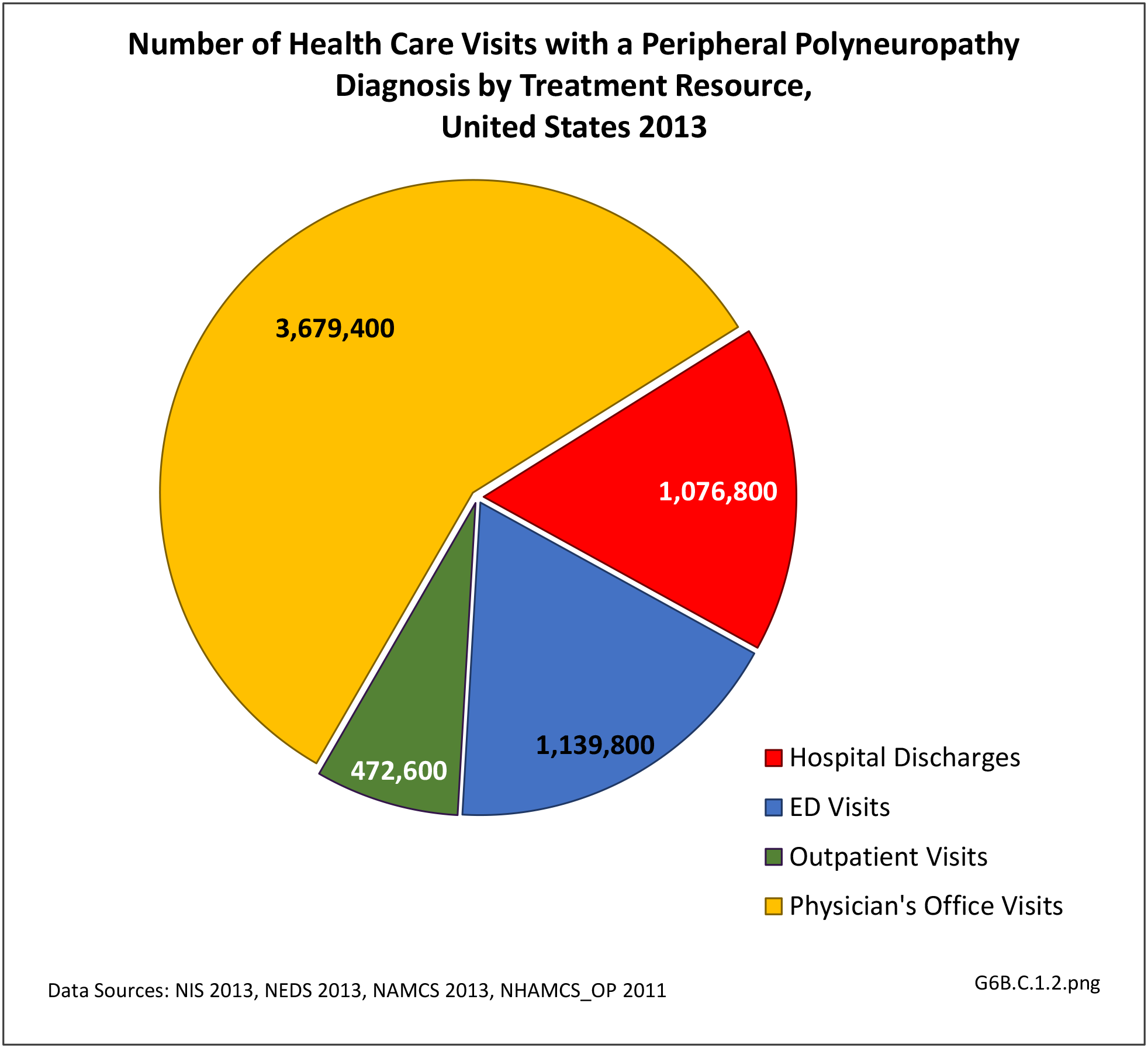 [23]
[23] The prevalence of peripheral neuropathy increases with age1 and the underlying etiology is diferent among different age groups. In general, manifestations of peripheral neuropathy tend to be severe in the elderly. Diabetic peripheral neuropathy is a significant contributor to falls, fall-related injuries, and overall impaired mobility, compounding the normal decline seen with aging.2 It also impairs activities of daily living and quality of life in the elderly.3
Direct medical costs related to peripheral neuropathy includes costs for diagnostic procedures (EDx, blood, genetic tests, etc), prescription medications (especially for pain medication), rehabilitation costs (physical and occupational therapy and bracing), and costs for complications related to peripheral neuropathy like fractures, non-healing wounds, etc. According to a commercial claims database, there was a 46% increase in the annual cost per patient associated with visits to hospitals, emergency departments, doctors' offices and pharmacy claims after diabetic peripheral neuropathy was diagnosed. The greatest cost increase was associated with hospitalization.1
In 2013, mean hospital charges for discharges associated with peripheral neuropathy were $50,500 for an average stay of 5.8 days. Hospital charges, which totaled $54.4 billion, are not the actual cost due to differences in payment structures, plus the additional cost of professional fees and associated treatments noted above. In addition, nearly half (48%) of hospital discharges were discharged to additional care such as inpatient rehabilitation or skilled nursing facilities. Patients initially seen in an emergency department were, more often than not (61%), admitted to the hospital. (Reference Table 6B.2.1 PDF [16] CSV [17]and Table 6B.2.2 PDF [18] CSV [19])
Motor neuron diseases (MNDs) are a group of rare disorders affecting motor neurons (nerve cells) that transmit signals from the brain to the muscles in the body. They present with muscle weakness and wasting, resulting in impaired walking, fine motor skills, limitations in activities of daily living, swallowing, speech, and eventually breathing. Motor neuron diseases can be classified into acquired (non-inherited) or inherited. There are no direct tests to identify MNDs, with diagnosis often the result of ruling out other conditions that early symptoms can mimic. In addition, there is no cure or standard treatment for MNDs. Generally, treatment consists of addressing symptoms, compensating for impairments, and providing palliative and supportive care. Some MNDs stabilize for long periods of time, while some rapidly progress to death in a few years.
Amyotrophic lateral sclerosis (ALS), often known as Lou Gehrig’s disease, is the most common type of motor neuron disease. It is usually rapidly progressive, has an unclear cause, and lacks a definite cure. According to the ALS registry, prevalence is 4 to 5 per 100,000 people, affecting more than 13,000 people1 Prevalence has been increasing over time due to better identification of cases. It is a fatal condition with short life expectancy after diagnosis.
The diagnosis of MND is made from the physician's interpretation of symptoms, while using selective diagnostic tests to confirm the diagnosis and rule out other mimicking conditions like spinal stenosis, cervical myelopathy or peripheral neuropathy. Diagnostic tests may include an EDx evaluation, lumbar puncture and MRIs of the spine and occasionally the brain. As there is currently no definitive cure for motor neuron disease, supportive care and prevention of unnecessary complications is the mainstay of management. Supportive care includes physical, occupational and speech therapy, respiratory care, nutritional and psychological support. Interdisciplinary care in a specialized center has been shown to provide superior care with slightly better survival in patients with ALS.1 Care is provided primarily at outpatient clinics, however, patients with debilitating symptoms require hospital admissions that are often lengthy and costly.2 Palliative care is required at the advanced stages of ALS.
In 2013, 74,200 health care visits included a diagnosis of a motor neuron disorder. Because of the small number, the specific cause of the disorder could not be identified, and only hospital discharges and ED visits were of sufficient size to be included. An equal number of males and females were included, with the diagnosis more common among the aging population. Those 65 and over accounted for 60% of motor neuron disorder diagnoses, and ages 45 to 64 another 30%. Only hospital discharges were available by race/ethnicity, with non-Hispanic whites accounting for 74% of the diagnoses, compared to being 62% of the population. Geographic region in the US was not a major factor. (Reference Table T6B.1.1 PDF [2] CSV [3]; Table 6B.1.2 PDF [4] CSV [5]; Table 6B.1.3 PDF [6] CSV; [7] and Table 6B.1.4 PDF [8] CSV [9])
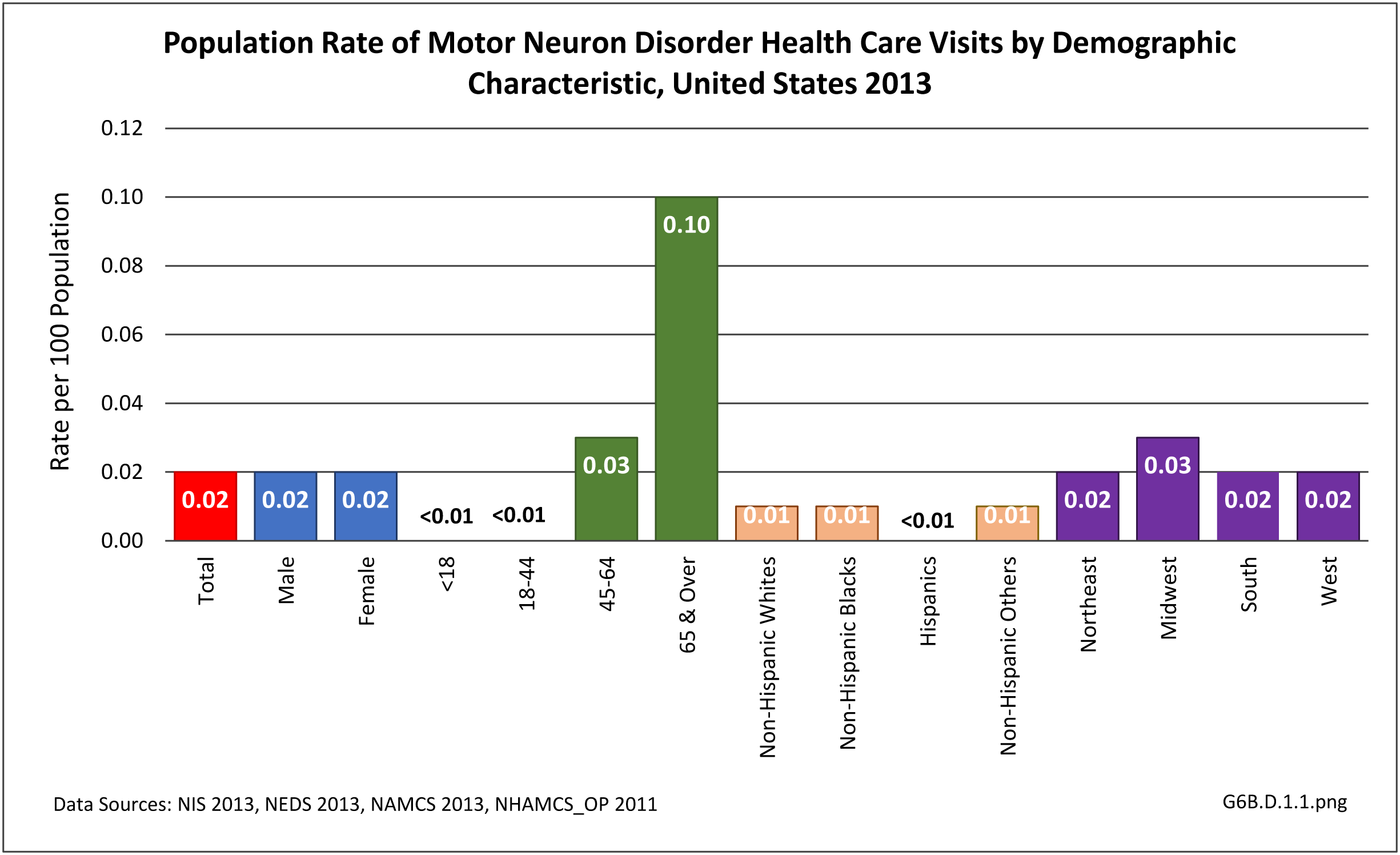 [25]
[25] As previously noted, only discharges/visits from/to the hospital and emergency department had sufficient numbers to be analyzed; the two sites were evenly represented. (Reference Table 6B.1.5 PDF [12] CSV [13])
Incidence rates of ALS increase with age, peaking between 70 and 80 years. Amyotrophic lateral sclerosis is a fatal condition for most patients with mean life expectancy of about 3 years after diagnosis, although some patients live longer.
Poliomyelitis is a viral infection affecting the nervous system which causes motor neuron disease. Although it has been eradicated by vaccines developed in the 1950's, polio survivors can suffer from post-polio syndrome for several decades (mean of 36 years) with gradual weakness, fatigue and muscle wasting. A study published in 1994-1995 estimated there were about 1 million polio survivors in the U.S., with 443,000 reported having had paralytic polio. Considering 25 to 40 percent of polio survivors develop the post-polio syndrome, it is one of most prevalent motor neuron disease in the elderly population.1
It is difficult to estimate the economic costs related to motor neuron disorders due to their mixed presentation. However, due to the seriousness of these conditions, and the need for a broad range of treatments and supportive care, the burden of MNDs is significant compared to their prevalence. Nearly two-thirds (58%) of hospital discharges with a diagnosis of MND were discharged to additional care. Mean hospital charges for an average stay of 6.1 days were $55,000 (Reference Table 6B.2.1 PDF [16] CSV [17] and Table 6B.2.2 PDF [18] CSV [19])
Myopathies are a group of primary muscle disorders causing muscle weakness, occasionally stiffness, and rarely muscle pain. They can be classified into inherited or acquired myopathy. Acquired myopathy can be further divided into idiopathic (cause unknown), infectious, metabolic, inflammatory, endocrine, and drug induced based on the etiology. Depending on the involvement, the patient can present with difficulty with mobility, including difficulty with rising from a chair, stair negotiation, or walking, and impaired activities of daily living, such as dressing and personal care due to difficulty raising the arms. Sensation is not primarily affected although the pain is not uncommon.
Inherited Myopathy
Inherited myopathy has a genetic basis and is passed from parent to child. The muscular dystrophies are the most well-known form of inherited myopathy, with progressive weakness and degeneration of the muscles controlling movement. Duchenne muscular dystrophy is the most common form of muscular dystrophy, primarily affecting 1 boy in 3,300, and is caused by absence of dystrophin, a protein in the muscle cell membrane. Other muscular dystrophies include Becker muscular dystrophy, which is less severe than Duchenne MD; fascioscapulohumeral muscular dystrophy (FSH, FSHD), in which the muscles of the face, shoulder blades, and upper arms are most affected; and myotonic muscular dystrophy, characterized by progressive muscle wasting and weakness with prolonged muscle contractions (myotonia) and inability to relax certain muscles after use.
Acquired Myopathy
Inflammatory myopathy, the most common acquired myopathy, includes a group of myopathies with chronic muscle inflammation marked by weakness and, occasionally, muscle pain. It is reported to occur in 8.9 in 1,000,000 persons, with prevalence gradually increasing over the time due to improved diagnostic techniques.1 Other forms of acquired myopathy can be secondary to infection, drugs ortoxins, or systemic (typically endocrine) disorders. Polymyositis, dermatomyositis, and inclusion body myositis are the common inflammatory myopathies.
Neuromuscular Junction Disorders
Myathenia gravis is a disorder affecting neuromuscular junctions, the contact points between the muscles and nerves. It presents with fatigue, fluctuating weakness, typically affecting the muscles that control eye and eyelid movement, chewing, and talking, resulting in drooping of the eyelid (ptosis), difficulty swallowing (dysphagia) or speaking (dysarthria).
Patients with myopathy are likely to utilize ambulatory visits, specialist visits, and hospitalization more often than those without myopathy, according to a study of the patients with inflammatory myopathy in a large managed care system.1 Diagnostic tests, including EDx, blood and genetic testing and muscle biopsy, are often utilized to confirm clinical suspicions from history and physical examination, and to delineate the exact type and underlying etiology of myopathy. As cure is limited in most myopathies, treatment focuses on the supportive and symptomatic treatment. In the initial stages and/or milder forms of myopathy, treatment is usually at an outpatient clinic for physical, occupational, and speech therapy, and bracing if needed. In progressive and disabling myopathies, hospitalization is required to manage the secondary problems related to the myopathies such as respiratory failure.
There were 792,700 health care visits made in 2013 with a diagnosis of myopathy or neuromuscular junction disorder. As with some other neuromuscular conditions, the overall number of visits to outpatient clinics and physician’s offices were too small to include in demographic analysis, and affect rates of incidence by demographic characteristics except for males and persons age 44-65 visits to outpatient clinics. Gender and geographic region are not factors, but as with other neuromuscular conditions, the condition increases with age. Racial differences are difficult to determine due to small sample sizes and unreliable data. (Reference Table T6B.1.1 PDF [2] CSV [3]; Table 6B.1.2 PDF [4] CSV [5]; Table 6B.1.3 PDF [6] CSV [7]; and Table 6B.1.4 PDF [8] CSV [9])
 [29]
[29] More than half (56%) of 792,700 2013 health care visits with a diagnosis of myopathy or neuromuscular junction disorder were to a physician’s office; however, data was not reliable at the level of sub-category conditions. The remainder of visits were spread between hospital discharges (13%), ED visits (13%), and outpatient clinics (18%). Acquired myopathy was the most common diagnosis with a hospital discharge, while neuromuscular junction disorder was most common in the ED.most common in the ED. (Reference Table 6B.1.5 PDF [12] CSV [13])
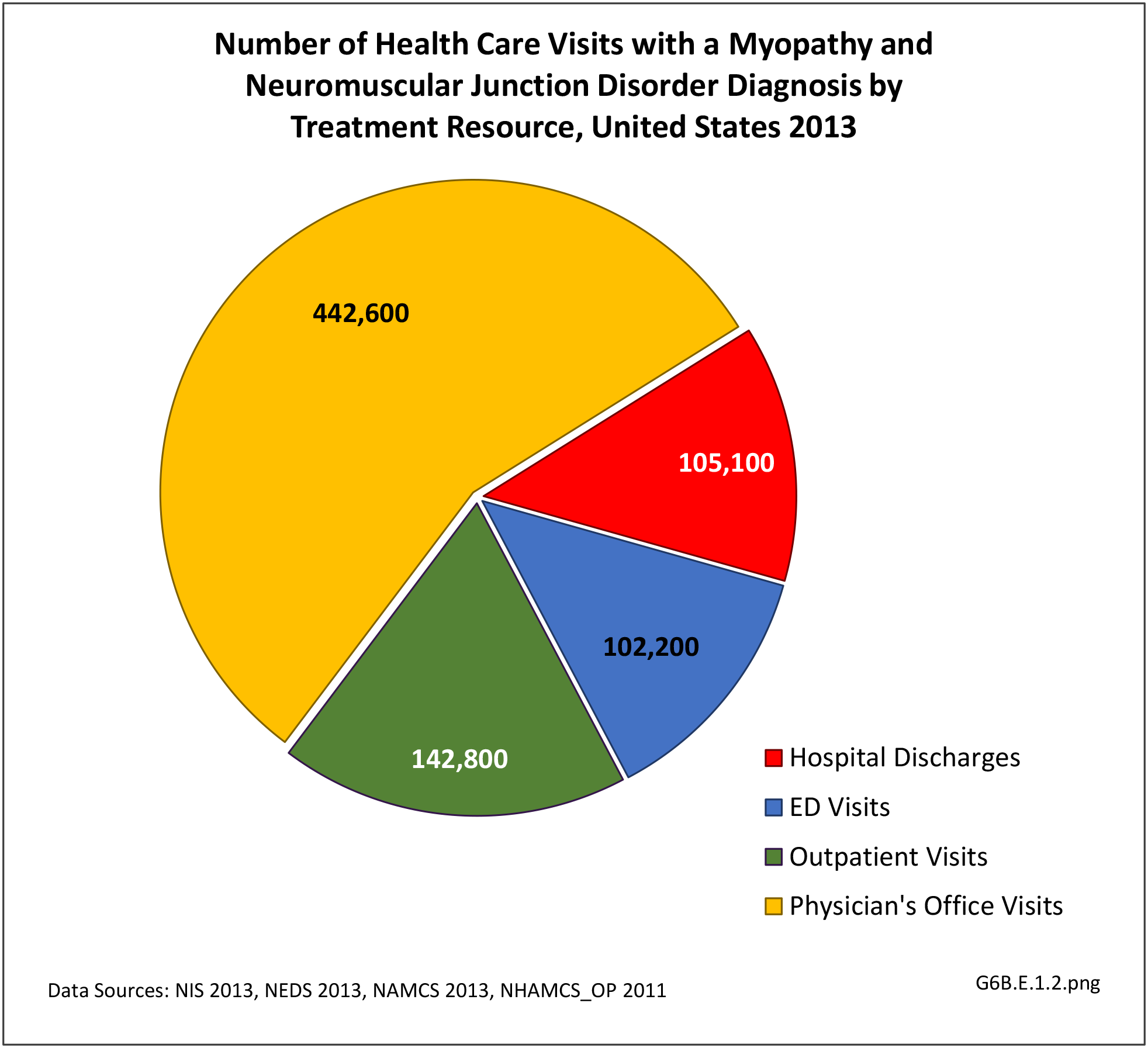 [31]
[31] Some myopathies are more common in the elderly, such as inclusion body myositis and myopathy secondary to cholesterol lowering medication (statin myopathy).
Patients with myopathies survive longer with better care and the increasingly recognized negative impact of aging on physical function and quality of life. In addition to age associated loss of muscle mass and strength (sarcopenia), muscle weakness and fatigue from myopathy can limit independence due to mobility and ADL issues.1
Studies of health care costs and resource utilization in patients with inflammatory myopathy in managed health care revealed annual medical costs were higher among newly diagnosed patients and those with existing inflammatory myopathy compared to unaffected controls.1
The direct annual medical cost of Duchene's muscular dystrophy (MD) ranges from $20,000 to over $50,000 depending on the study methodology. Medical costs include direct cost related to the myopathy and secondary problems including cardiac, respiratory, nutritional and spine complications. Medical costs related to MD and its secondary problems both increase over time.2
Mean hospital charges in 2013 for the diagnosis of myopathy was $81,800 for a mean stay of 8.9 days. This was the longest stay and highest mean charges of all neuromuscular disease categories. (Reference Table 6B.2.1 PDF [16] CSV [17] and Table 6B.2.2 PDF [18] CSV [19])
Spinal cord injury (SCI) is damage to the spinal cord, the bundle of nerves running from the base of the brain (brainstem) to the upper part of the lumbar spine. SCI disrupts communication between the brain and the rest of the body below the level of the injury, and depending on the severity resulting in the inability to move limbs, loss of sensation, bowel and bladder function. Depending on the underlying mechanism of injury, SCI can be divided into traumatic and non-traumatic causes. It can be further classified by the level of injury: tetraplegia involving all four limbs or paraplegia involving legs only; and the severity of injury: complete vs incomplete, with incomplete tetraplegia being most common.
Traumatic Spinal Cord Injury
Significant trauma to the vertebral column encasing the spinal cord can result in spinal cord injury. In a person with a vulnerable bony spine, for example someone with osteoporosis or ankylosing spondylitis, weakness in the supporting structure, such as with rheumatoid arthritis or Down's syndrome, or narrowing of the spinal canal due to spinal stenosis, a minor trauma or injury can result in spinal cord injury. The common underlying cause of injuries include motor vehicle accidents, followed by falls, violence such as gunshot wounds or assault, sports injuries, and industrial accidents. The A [32]merican Spinal Injury Association (ASIA) scoring system [33] is widely utilized by healthcare providers for further classification of SCI based on the injury level and severity.
Non-traumatic spinal cord injury
Spinal cord injury also can be secondary to multiple sclerosis (MS), inflammatory conditions, compression by bony spurs or herniated discs, and metastatic cancer, all disrupting spinal cord function. MS is a central nervous system (brain and spinal cord) disorder that damages the myelin sheath surrounding the nerve cells and fibers, and can presents with symptoms of spinal cord dysfunction, as well as disruption of vision, speech or cognitive function. A condition known as transverse myelitis is an inflammation across both sides of one level, or segment, of the spinal cord resulting in temporary or permanent symptoms that include paralysis and loss of sensation, bowel and bladder control. The segment of the spinal cord where the damage occurs determines the parts of the body affected, much like with a traumatic SCI.
SCI is a life changing event affecting a younger population (average age at injury: 42 years old) and it is a cause of major disability. Annual incidence of spinal cord injury is approximately 54 cases per million in the US, with approximately 17,000 new cases of SCI each year. The prevalence is estimated to be 282,000 persons alive with a SCI in 2016.1 Patients are initially admitted to acute care units of hospitals for stabilization for an average length of stay of 11 days followed by inpatient rehabilitation with an average 35 day length of stay.1,2 As cure is limited in most cases of SCI, patients require continuous outpatient care including intermittent physical, occupational and speech therapy, pain management, and prevention of complications directly or indirectly related to SCI including deep vein thrombosis, pressure ulcers, pneumonia and urinary tract infections.
Males account for approximately 80% of new SCI cases each year, with nearly 1 in 4 (22%) injuries occurring to non-Hispanic blacks since 2010, nearly twice the proportion of non-Hispanic blacks in the general population (12%).2
Analysis by demographic variables is limited by data size for outpatient clinic and physician’s office visits. However, as previously noted, males have a higher rate of SCI health care visits, and age is clearly a factor beginning in middle age around 45. Geographic region does not appear to be a factor. Race/ethnicity is unclear due to missing data cells. However, with a rate of 0.30 per 100 persons compared to 0.25 for all races, SCI health care visits appear to be greater among non-Hispanic whites than in other races/ethnicities. (Reference Table T6B.1.1 PDF [2] CSV; [3] Table 6B.1.2 PDF [4] CSV [5]; Table 6B.1.3 PDF [6] CSV; [7] and Table 6B.1.4 PDF [8] CSV [9])
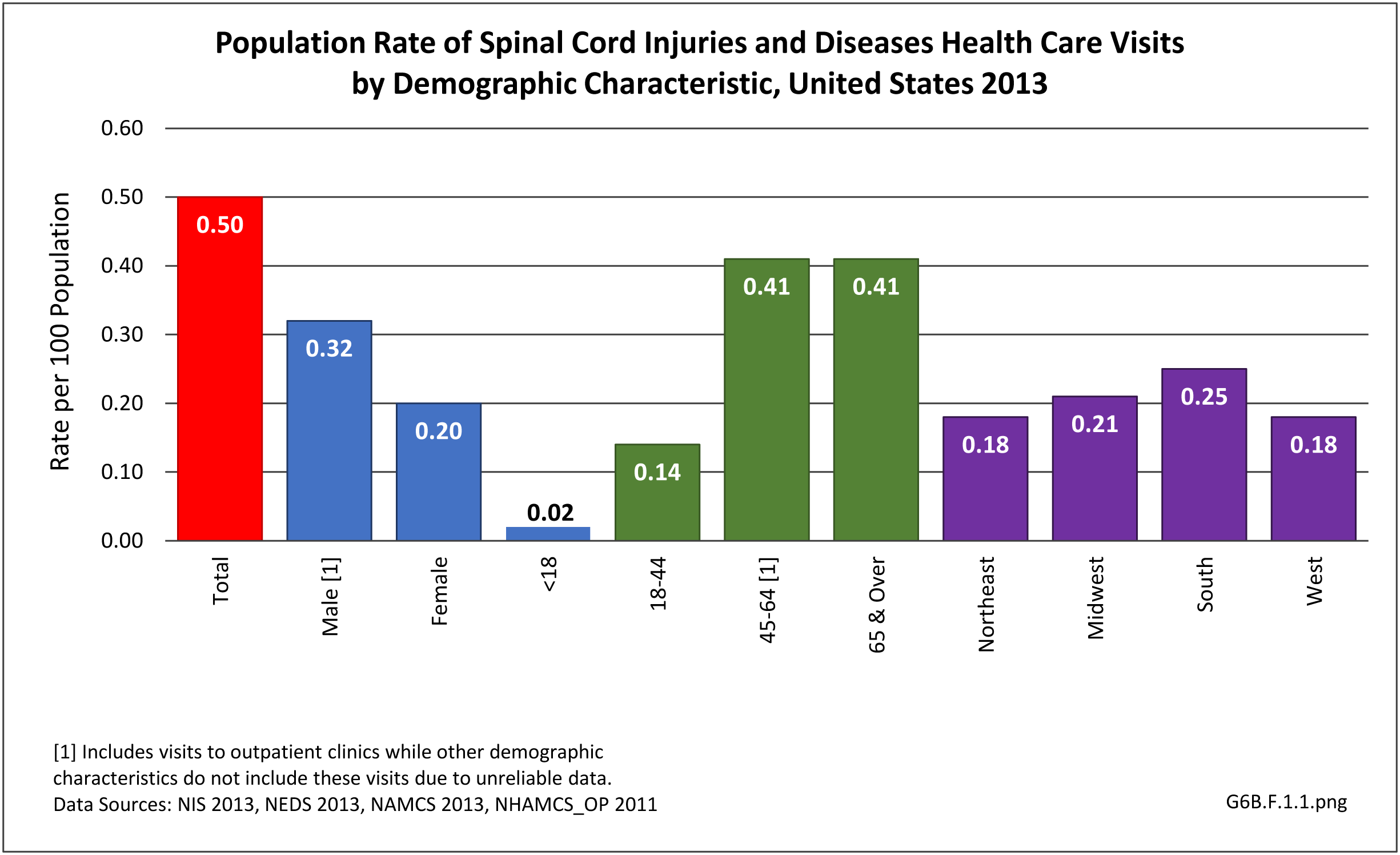 [35]
[35] In 2013, spinal cord injury or disease was diagnosed in 1.56 million health care visits, representing 1 person in every 200 in the US. However, it is likely more than one visit per person would reduce this ratio. Visits to outpatient clinics and physician’s offices generally do not meet standards of reliability, but for hospital discharges and emergency department visits with a diagnosis of spinal cord injuries and diseases, a diagnosis of ‘other paralytic syndromes’ account for 75% of visits. Nearly half (47%) of visits are to physician’s offices, while 1 in 5 (20%) involves hospitalization. (Reference Table 6B.1.5 PDF [12] CSV [13])
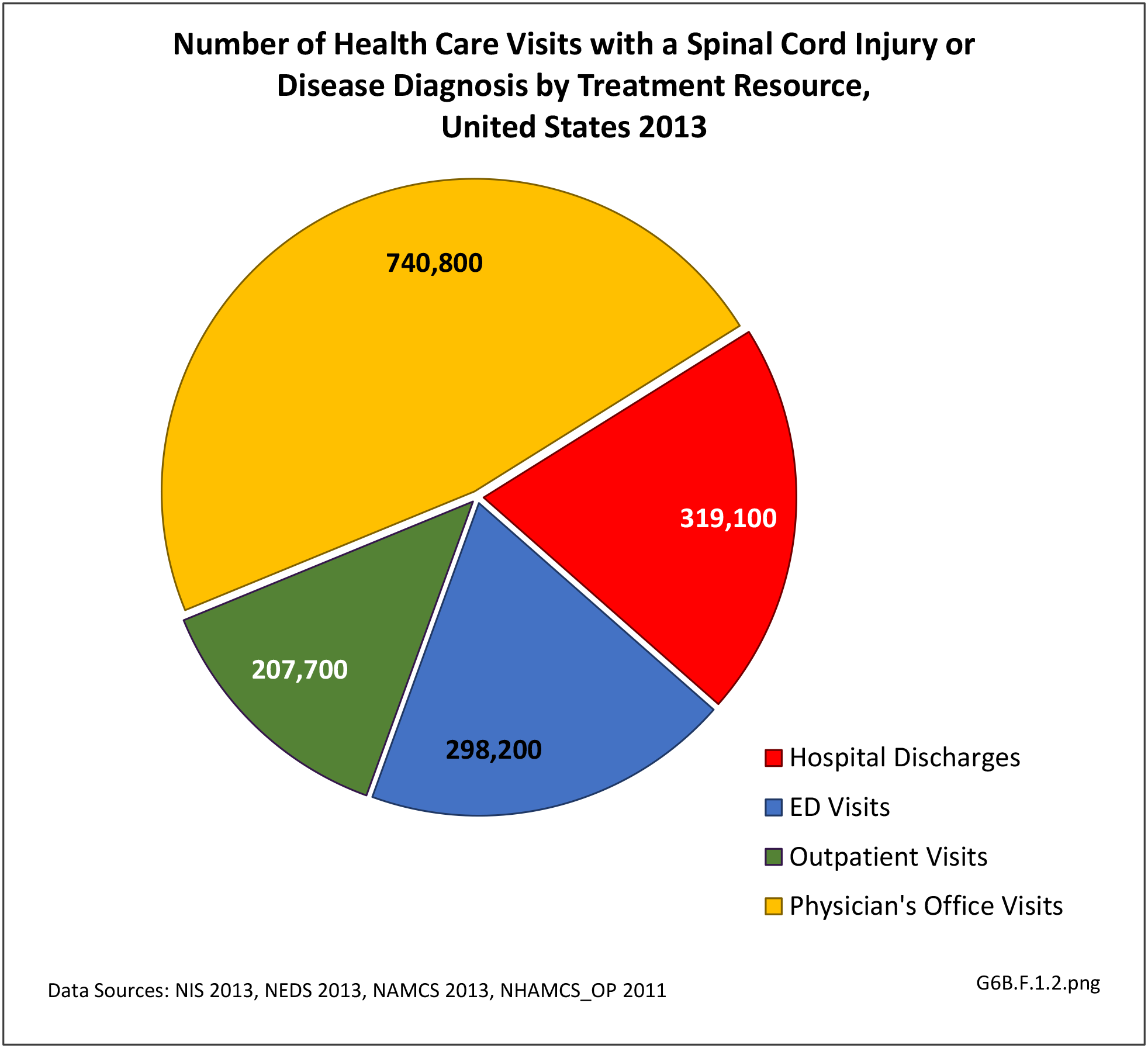 [37]
[37]
The mean age at the time of spinal cord injury increased from 29 years in the 1970s to 42 years in 2016.1 Non-traumatic spinal cord injuries are increasing, in part, due to the aging of the population and the concurrent age-related health conditions with a greater likelihood of minor events causing a SCI, like falls in an arthritic or stenotic spine. There is a possibility non-traumatic SCI will surpass the incidence of traumatic SCI in the future.2 The lifespan for SCI survivors has not changed since the 1980’s, and remains significantly shorter than for healthy counterparts. In 2016, for persons who survived the first 24-hours post injury, life expectancy for a 20-year old with the lowest level SCI is 52.6 years and for a 20-year old with a high tetraplegia 35.7 years. Comparable life expectancy for a 60-year old at time of injury is 17.9 years for a low level injury and 8.1 years for a high level tetraplegia, respectively.1 SCI survivors report more health problems, with significant impacts on physical function and quality of life posing greater challenges in aging SCI survivors.3
Mean hospital charges in 2013 for hospital stays with a diagnosis of spinal cord injury or disease were $80,700, with a mean stay of just over 8 days. Total hospital charges in 2013 were $25.7 million. Due to the severity of spinal cord injuries and diseases, this is only a small part of overall health care costs that usually last a lifetime. (Reference Table 6B.2.1 PDF [16] CSV [17]and Table 6B.2.2 PDF [18] CSV [19])
The average expense for the first year of injury in 2015 dollars, including health care costs and living expenses, ranges from $347,900 for the lowest level of injury to $1.1 million for a high tetraplegia. Average yearly expenses for each subsequent year of survival range from $42,300 to $185,100. These costs do not include indirect costs such as lost wages, fringe benefits, and productivity, which were estimated to be over $72,000 per year.1 In addition, there is significant economic and emotional burden to spouses and other family members.
The overall burden of all neuromuscular conditions is difficult to assess due to the life-long care usually required following onset of the disease. However, comparison of the 1.9 million hospital discharges and 2.4 million emergency department visits for all health conditions provides some sense of the magnitude of burden.
Neuromuscular conditions were 5% of all hospital discharges and 2% of all emergency department visits in 2013. Discharge from the hospital to another form of care (short/intermediate/nursing home facility or home health care) occurred in 47% of discharges with a neuromuscular diagnosis, compared to 27% of discharges for all health care reasons. Among ED visits, 46% were admitted to the hospital when there was a neuromuscular diagnosis, compared to 14% for all health care reasons. Thus, it can be seen than neuromuscular conditions have a much higher level of long-term care than health conditions overall. (Reference Table 6B.2.1 PDF [16] CSV [17])
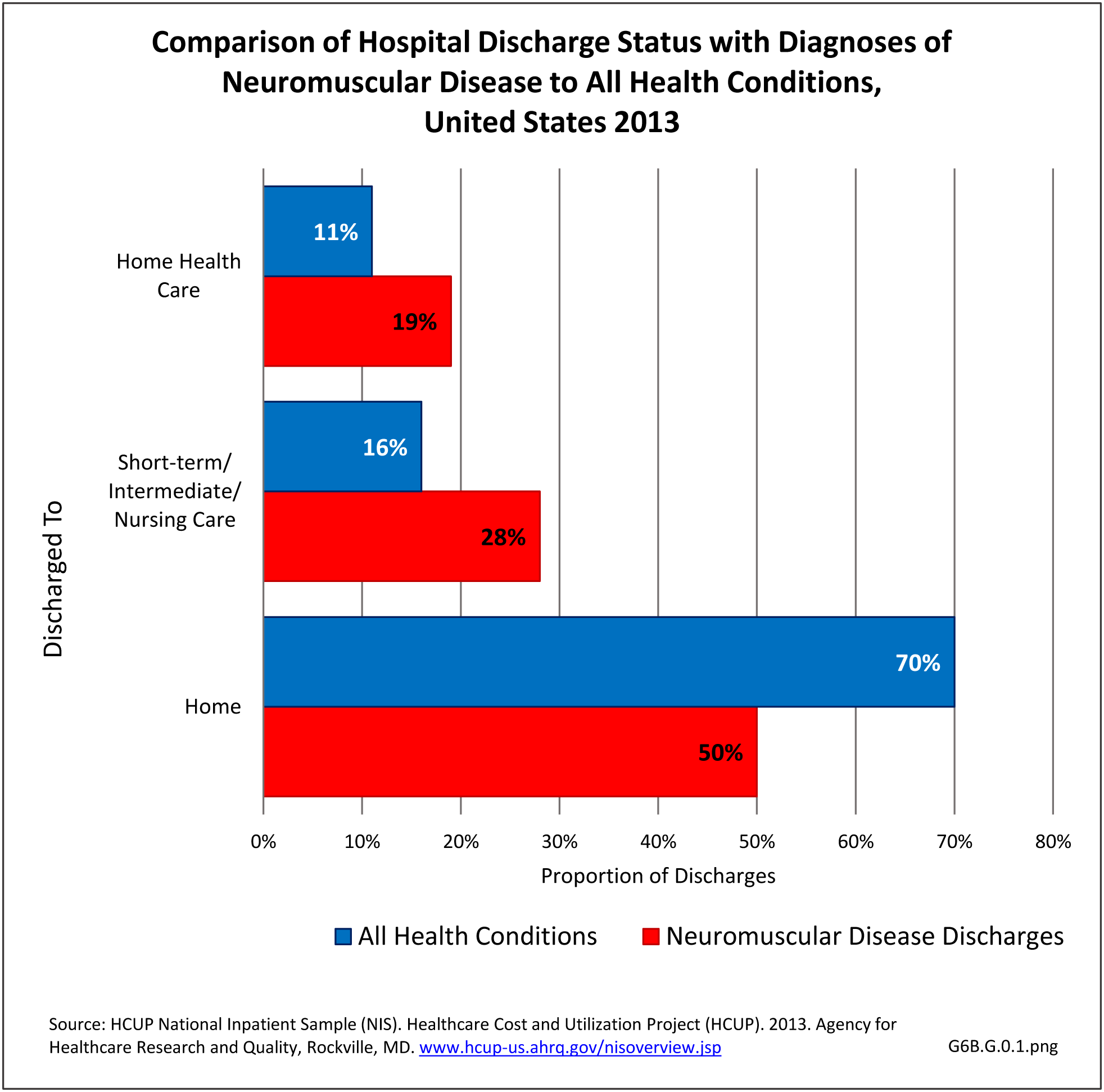 [40]
[40] Mean hospital charges in 2013 for all discharges with a neuromuscular disease diagnosis were $59,100, 38% greater than mean charge for discharges with any health condition. Mean length of stay for persons with a neuromuscular disease were 26% longer than the mean stay for all hospitalizations (5.9 days versus 4.7 days. Total hospital charges for neuromuscular diseases in 2013 were $109.8 million, 7.2% of total hospital charges for all health conditions, even though discharges for a neuromuscular disease comprised only 5% of all hospitalizations. (Reference Table 6B.2.2 PDF [18] CSV [19])
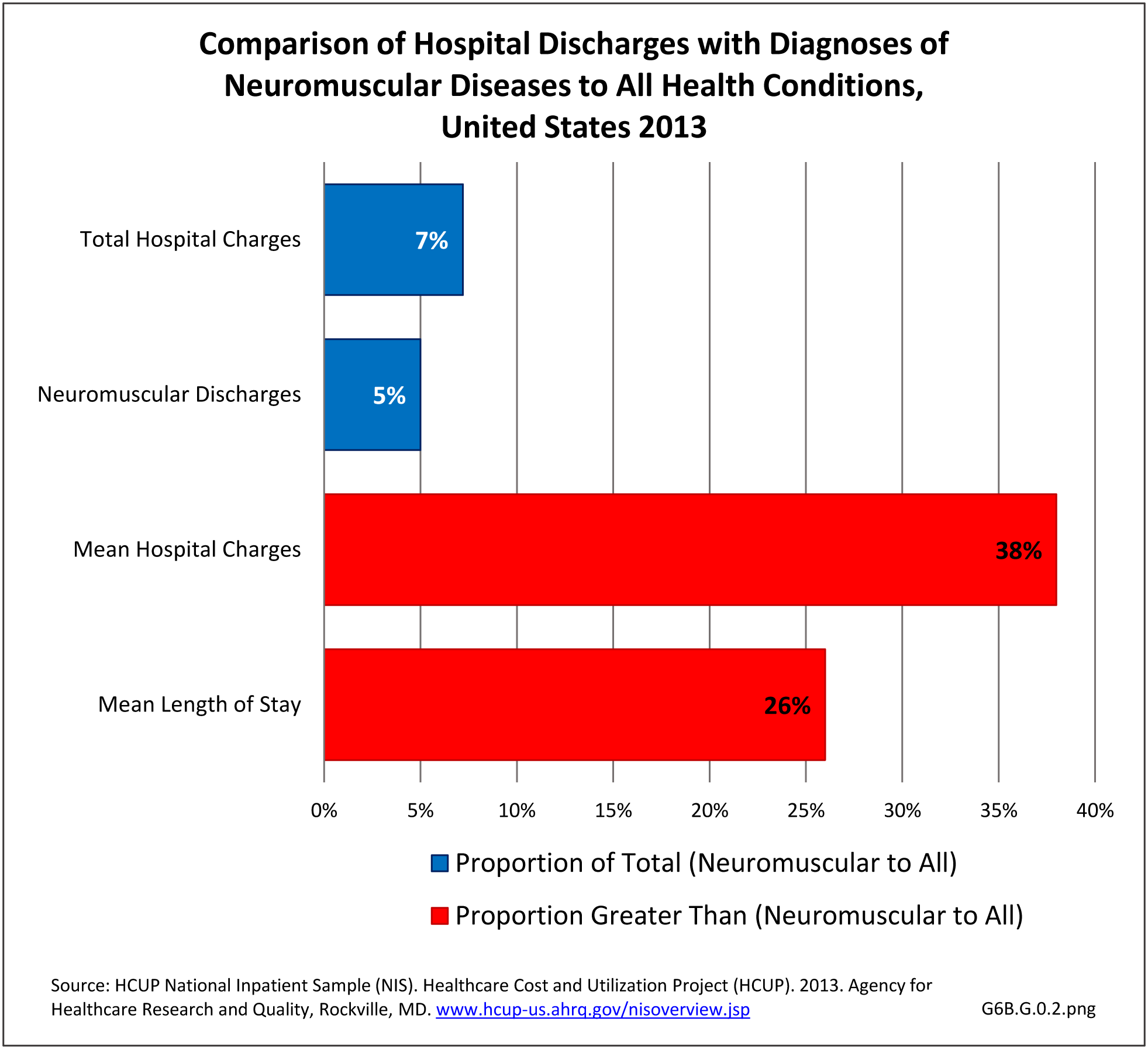 [42]
[42] As discussed above, neuromuscular diseases become more prevalent as the population ages. The primary exception to this is spinal cord injuries or diseases, which occur in younger populations more than in the elderly. Total health care visits with a neuromuscular diagnosis reflected this trend in 2013, particularly in the middle age range of 45 to 64. Since many neuromuscular diseases are life-long conditions once they occur or are diagnosed, care will be ongoing throughout life.
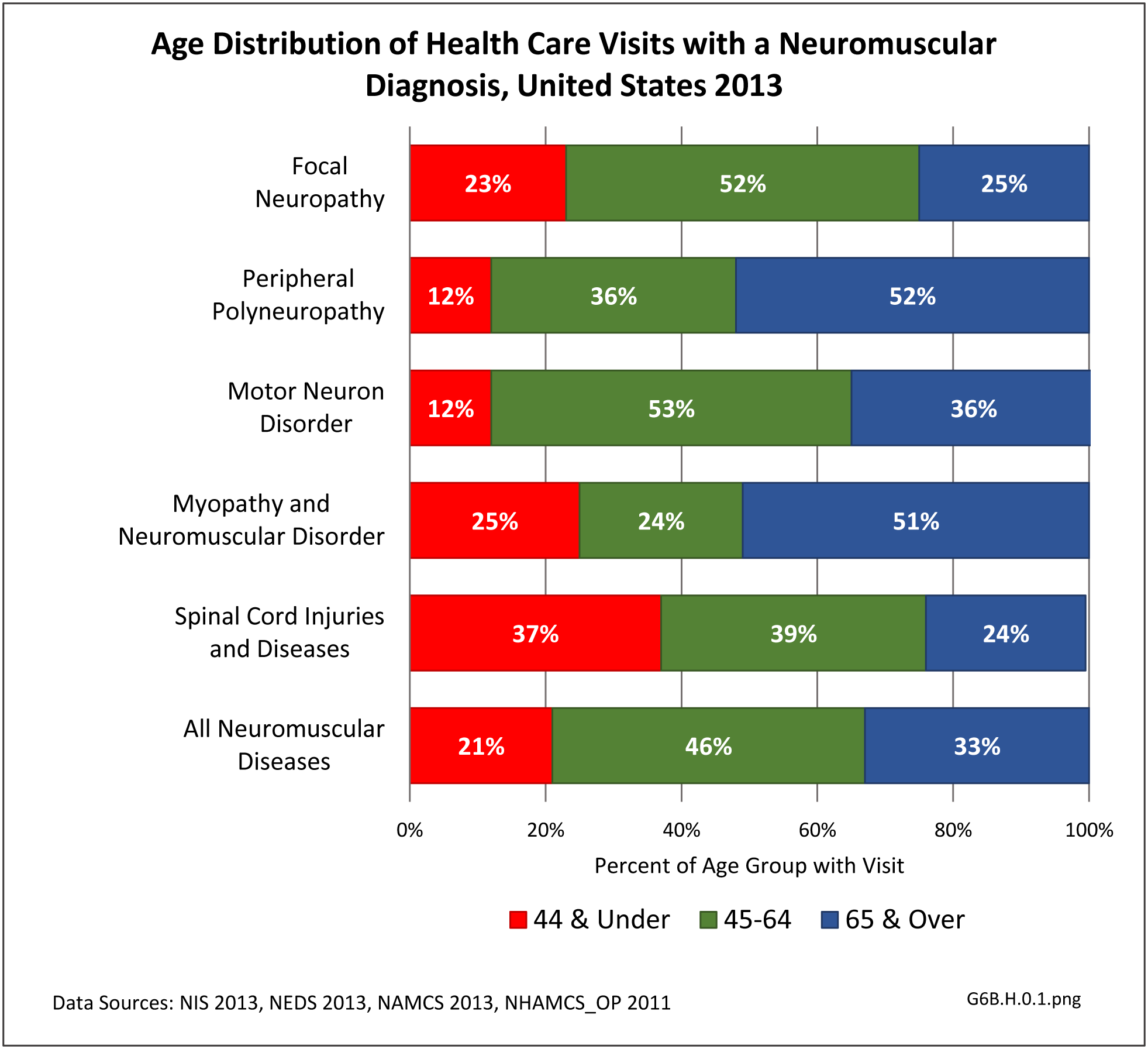 [44]
[44] The true prevalence and burden of neuromuscular diseases is likely underestimated due to insufficient research in the area. These conditions often cause significant pain, motor impairment, loss of work and can lead to chronic disability. They may require lifelong rehabilitative care, utilizing many resources in the form of pain management, physical and occupational therapy, bracing, wound and nursing care. Quality of life can be severely affected.
These are an important group of diseases not only because of their direct impact, but also their indirect impact leading to other musculoskeletal conditions such as accelerated degenerative joint disease, scoliosis and osteoporosis. They have a high caregiver burden, and often lead to emotional strain on patients and families.
The datasets assessing hospital discharges are compelling in that patients with neuromuscular disease stay in the hospital longer, at a higher cost, and are discharged to places other than home more often. This does not tell the whole story, however, as many of these conditions are treated in the outpatient setting. Relying on diagnosis codes can also underestimate prevalence since many patients are admitted or treated in an outpatient setting based on symptoms, without a clear diagnosis. Often the diagnosis is clinical, and the correct diagnostic code not used.
There is a scarcity of research on cost effective health care model systems for those with neuromuscular disease despite increasing direct and indirect financial burdens on patients and their families. Despite the recent introduction of the patient-centered outcomes research institute,1 there is a shortage of outcome based research from the neuromuscular patient perspective.
Current postgraduate education and training of clinicians is limited in the management of chronic neuromuscular disease. This includes prevention of complications (for example, prevention strategies for falls specific to this population), and cost effective management at the community level (for example, home based interventions using information technology/telemedicine). There is a lack of training and resources available to primary care providers who manage patients with neuromuscular disease in typical community settings. The ever increasing demand on physicians to see high volumes of patients may also be impacting the quality of evaluations of neuromuscular diseases which typically require intense and lengthy historical intakes. Collaboration amongst interdisciplinary teams, rehabilitation specialists, health care leadership, public health stakeholders, researchers, and academic institutions to educate future healthcare providers in neuromuscular disease care could be improved.
One of the key challenges in the care of people with neuromuscular disease is that patients are living longer with these often chronic diseases. As other disabling musculoskeletal and non-musculoskeletal disorders occur in this population, it can be harder for individuals to maintain quality of life and independence. Pain management for patients with neuromuscular diseases can be challenging, especially with the current opioid epidemic.1 Better biopsychosocial approaches to pain management need to be developed and implemented.
The increasing demand from the aging population with neuromuscular disease and a shortage of trained health care providers may place an excessive burden on communities. Furthermore, access to comprehensive care will be difficult for individuals with moderate to severe neuromuscular disorders requiring an interdisciplinary approach, especially in less populated areas. Controlling costs of medical care without compromising quality is another great unmet need, but not one unique to neuromuscular disease.
ICD-9-CM codes used in this analysis can be viewed by clicking here [45].
To view a crosswalk of ICD-9-CM codes with ICD-10-CM codes, click here [46].
Links:
[1] https://doi.org/10.2337/diaclin.23.1.9
[2] https://bmus.latticegroup.com/docs/bmus_e4_T6B.1.1.pdf
[3] https://bmus.latticegroup.com/docs/bmus_e4_T6B.1.1.csv
[4] https://bmus.latticegroup.com/docs/bmus_e4_T6B.1.2.pdf
[5] https://bmus.latticegroup.com/docs/bmus_e4_T6B.1.2.csv
[6] https://bmus.latticegroup.com/docs/bmus_e4_T6B.1.3.pdf
[7] https://bmus.latticegroup.com/docs/bmus_e4_T6B.1.3.csv
[8] https://bmus.latticegroup.com/docs/bmus_e4_T6B.1.4.pdf
[9] https://bmus.latticegroup.com/docs/bmus_e4_T6B.1.4.csv
[10] https://bmus.latticegroup.com/file/bmuse4g6bb11png
[11] https://bmus.latticegroup.com/docs/bmus_e4_G6B.B.1.1_0.png
[12] https://bmus.latticegroup.com/docs/bmus_e4_T6B.1.5.pdf
[13] https://bmus.latticegroup.com/docs/bmus_e4_T6B.1.5.csv
[14] https://bmus.latticegroup.com/file/bmuse4g6bb12png
[15] https://bmus.latticegroup.com/docs/bmus_e4_G6B.B.1.2_0.png
[16] https://bmus.latticegroup.com/docs/bmus_e4_T6B.2.1.pdf
[17] https://bmus.latticegroup.com/docs/bmus_e4_T6B.2.1.csv
[18] https://bmus.latticegroup.com/docs/bmus_e4_T6B.2.2.pdf
[19] https://bmus.latticegroup.com/docs/bmus_e4_T6B.2.2.csv
[20] https://bmus.latticegroup.com/file/bmuse4g6bc11png
[21] https://bmus.latticegroup.com/docs/bmus_e4_G6B.C.1.1_0.png
[22] https://bmus.latticegroup.com/file/bmuse4g6bc12png
[23] https://bmus.latticegroup.com/docs/bmus_e4_G6B.C.1.2_0.png
[24] https://bmus.latticegroup.com/file/bmuse4g6bd11png
[25] https://bmus.latticegroup.com/docs/bmus_e4_G6B.D.1.1_0.png
[26] https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2764727/
[27] https://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Post-Polio-Syndrome-Fact-Sheet
[28] https://bmus.latticegroup.com/file/bmuse4g6be11png
[29] https://bmus.latticegroup.com/docs/bmus_e4_G6B.E.1.1_0.png
[30] https://bmus.latticegroup.com/file/bmuse4g6be12png
[31] https://bmus.latticegroup.com/docs/bmus_e4_G6B.E.1.2_0.png
[32] http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3232636/
[33] https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3232636/
[34] https://bmus.latticegroup.com/file/bmuse4g6bf11png
[35] https://bmus.latticegroup.com/docs/bmus_e4_G6B.F.1.1_0.png
[36] https://bmus.latticegroup.com/file/bmuse4g6bf12png
[37] https://bmus.latticegroup.com/docs/bmus_e4_G6B.F.1.2_0.png
[38] https://www.nscisc.uab.edu/Public/Facts%202016.pdf
[39] https://bmus.latticegroup.com/file/bmuse4g6bg01png
[40] https://bmus.latticegroup.com/docs/bmus_e4_G6B.G.0.1_0.png
[41] https://bmus.latticegroup.com/file/bmuse4g6bg02png
[42] https://bmus.latticegroup.com/docs/bmus_e4_G6B.G.0.2_0.png
[43] https://bmus.latticegroup.com/file/bmuse4g6bh01png
[44] https://bmus.latticegroup.com/docs/bmus_e4_G6B.H.0.1_0.png
[45] https://bmus.latticegroup.com/docs/bmus_e4_ICD%209%20neureomuscular%20codes.pdf
[46] https://bmus.latticegroup.com/docs/ICD9%20%26%20ICD10%20neuromuscular%20codes.pdf